


The coverage of Q-Chem is wide, can estimate following ones.
- • Molecular structure
- • Chemical reaction
- • Molecular vibration
- • Electronic spectrum
- • NMR spectrum
- • Solvation effect
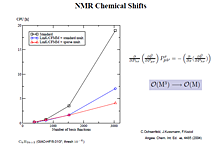
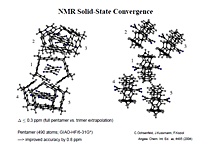

The new function of Q-Chem 6.0.1:
- • The DFT functional option which is added anew:
- º Long distance exchange interaction revision (LRC) functional
- º Baer-Neuhauser-Livshits (BNL) functional
- Variation of ωB97 functional
- º Restraint conditionality DFT (CDFT)
- º The empirical dispersion revision which is by Grimme
- • Solvation model
- º SM8 model for hydration and organic solvent (energy calculation analytical slope)
- º Update of Onsager reaction place model
- • Analysis of interaction between molecules:
- º SCF which uses the complete localization molecular track for molecular interaction (SCF-MI)
- º The Roothaan step which follows to SCF-MI (Roothaan-step) revision
- º Energy bisection (EDA)
- º COVP for charge-transfer (Complimentary occupied-virtual pair) analysis
- º BSSE which is automated (basis-set superposition error) calculation
- • Analysis of electronic movement
- • Relief restraint for SCF focus (Relaxed constraint) algorithm (RCA)
- • G3Large basis functional system for transition metal
- • New MP2 option:
- º The energy due to double basis type RIMP2 method analytical slope
- º O2 energy analytical slope
- • The calculation method where the wave function base in order to calculate the property of excitation state efficiently is new
- º SOS-CIS for excitation state (D) energy
- º SOS-CIS for excitation state (D0) energy and slope
- • Connection cluster method (Coupled-cluster methods):
- º IP-CISD energy and slope
- º EOM-IP-CCSD energy and slope
- º The parallel calculation in the connection cluster method which uses OpenMP
- • QM/MM method:
- º QM/MM complete Hessian appraisal
- º QM/MM MBH (mobile-block) hessian appraisal
- º Description of MM atom which uses the Gaussian nonlocalized electron
- • The vibrational analysis which uses Partial Hessian method
- • Wave function analytical tool:
- º Improvement of localization tracked calculation algorithm
- º Dispersion multipolar analysis
- º Analytical Wigner distribution function
